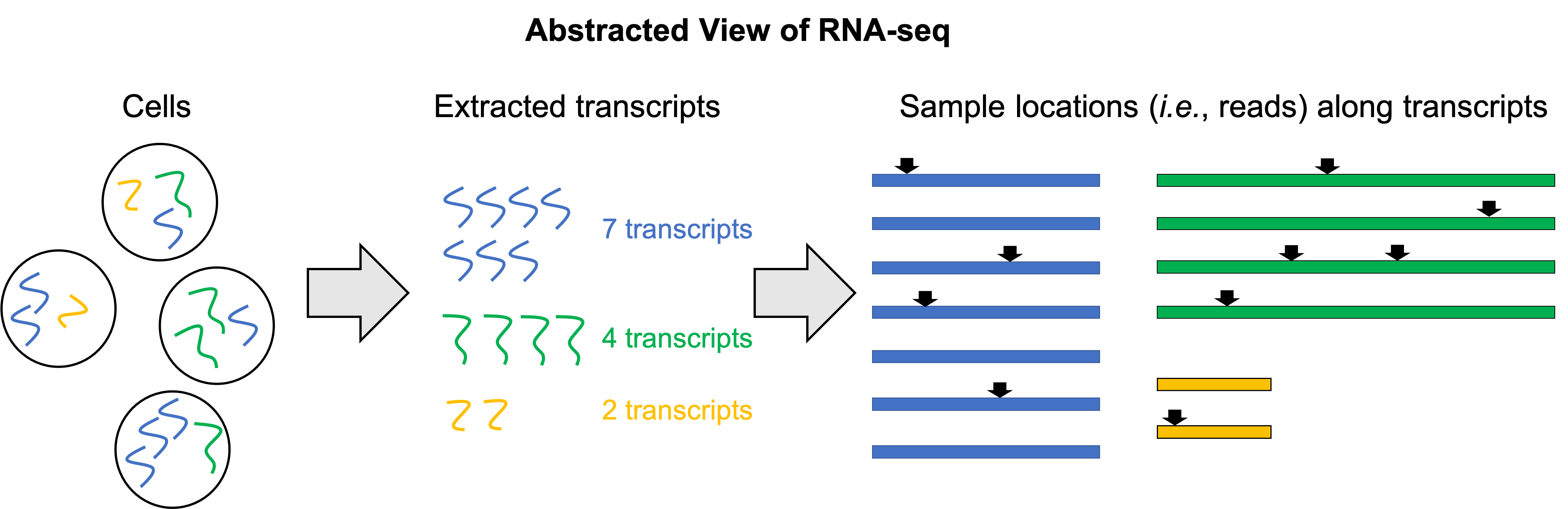
RNA-seq ( «secuenciación de ARN» ), también llamado Secuenciación del Transcriptoma Entero para Clonación al Azar[1] (del inglés Whole Transcriptome Shotgun Sequencing ), utiliza la secuenciación masiva (NGS) para revelar la presencia y cantidad de ARN, en una muestra biológica en un momento dado.[2][3]
Así, la RNA Seq usa para analizar cambios en el transcriptoma. Concretamente, la RNA-seq facilita la observación de transcritos resultantes del empalme alternativo, modificación postranscripcional, fusiones génicas, mutaciones/polimorfismos de nucleótidos únicos y cambios de expresión de genes.[4] Dado que el proceso implica la obtención de RNA total, la RNA Seq puede ayudar a caracterizar poblaciones diferentes de RNA como miRNA, tRNA, y rRNA[5] Esta tecnología, además, también puede servir para determinar las fronteras exón / intrón y verificar o enmendar regiones 5 'y 3'.
- Los datos obtenidos mediante RNA-seq han sido útiles para la realización de numerosos estudios. En un artículo publicado en 2019 en la revista Nature communications[6] se utilizan datos de ARN-seq para estudiar las firmas transcriptómicas de diferentes miembros del MTBC. Los resultados obtenidos en este estudio muestran que los diferentes clados MTBC tienen su propia firma transcriptómica.
Referencias
Enlaces externos